Interpreting PoET Results Table#
This tutorial explains how to interpret the results generated by the PoET Generate Sequences and Score Sequences tools.
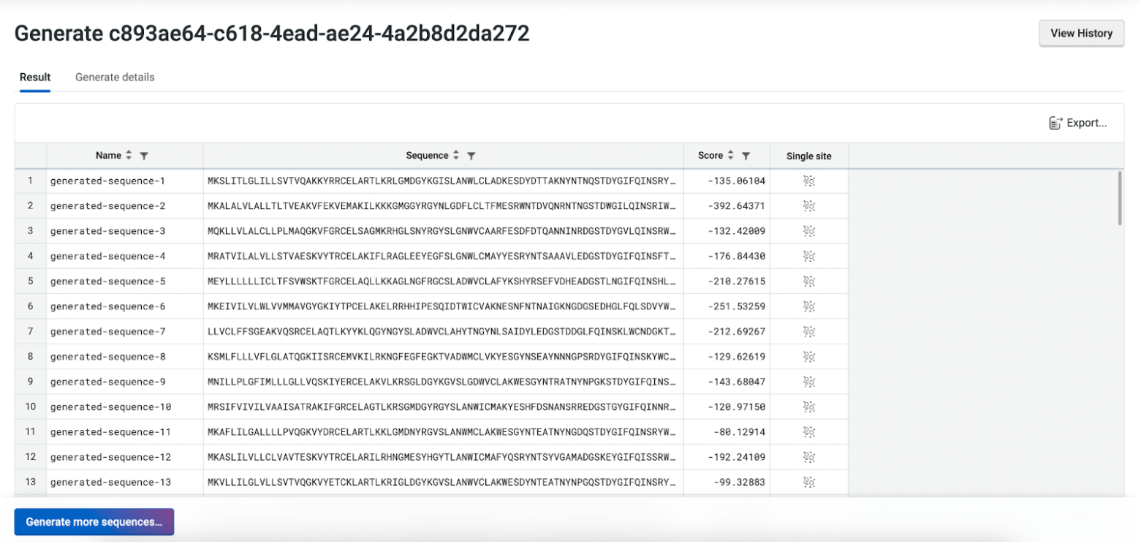
Results Table#
Your results are presented in a table, with each generated sequence assigned a log-likelihood score and other metrics, such as Identity, pLDDT, RMSD, TM Score, depending on the selected Alignment Method. You can customize the displayed columns using the right-click menu from the table header.
Right-click any sequence to assess local fitness landscapes through the Run Substitution Analysis menu. You can also sort, filter your results and save them to file using the Export button.
Structure Prediction and Comparison#
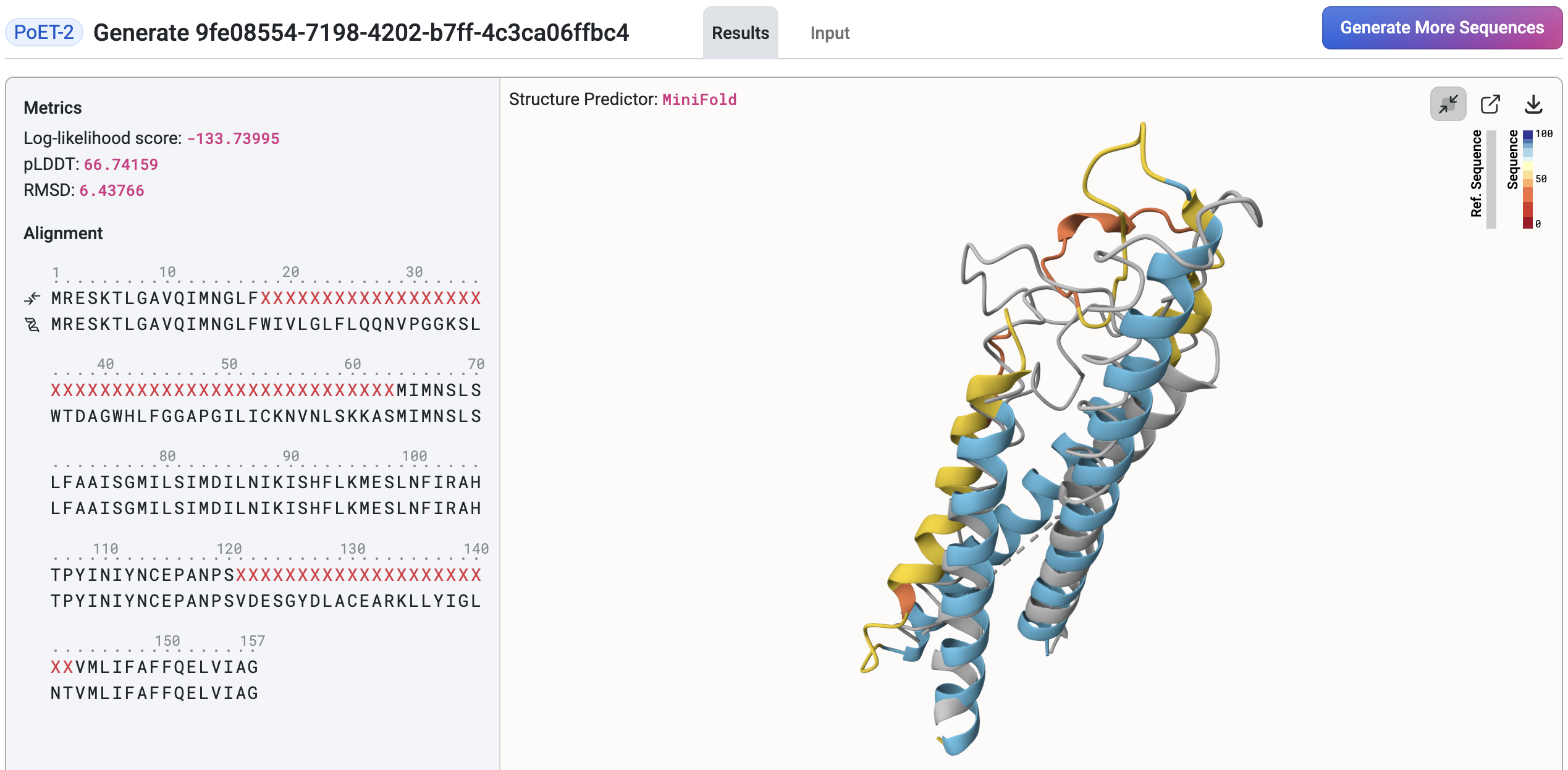
Once the structure prediction job completes, the structure viewer will appear on the right side of the page. The prediction model can be changed via the Structure Predictor dropdown located above the viewer. Hover over a sequence in the results table to preview and compare its structure against the query structure. The viewer also displays key metrics, including:
Log-likelihood score: Reflects how well the generated sequence fits the prompt. A higher score indicates a better fit.
Identity: The percentage of matching amino acids between the generated sequence and the reference across the aligned region.
pLDDT (predicted Local Distance Difference Test): A per-residue confidence score (commonly scaled 0-100) indicating how reliable each residue’s predicted position is. In this table, the value shown is the average pLDDT across residues, not per-residue scores.
RMSD (Root Mean Square Deviation): A measure of structural similarity between two molecules, typically comparing backbone atoms. Lower RMSD values indicate greater structural similarity.
TM Score (Template Modeling score): A measure of similarity between two protein structures (scaled 0-1), higher means more similar. Compared to RMSD, TM-score is more sensitive to global fold similarity than to local differences.
Click a sequence to expand the structure viewer, which will overlay the results table. In this view, you can examine detailed metrics and sequence-to-prompt alignment for the selected sequence.
Change Alignment Method#
The alignment method used for structural comparisons can be changed via the Alignment Method toggle buttons located above the results table. Available methods may include:
Structure: Structures are superimposed with TM-Align algorithm, and this superposition is used to compute structure similarity metrics, such as RMSD and TM Score.
Sequence: Sequences are aligned with Needleman-Wunsch algorithm, and then structures are superimposed via Kabsch algorithm using the sequence alignment. This superposition is then used to compute structure similarity metrics.
Off: If sequences are the same length, then they are aligned such that residues at the same position are aligned. Otherwise, no alignment is performed. In the event of a length mismatch, some metrics such as Identity, RMSD, and TM-score are not available.
Change Reference Structure#
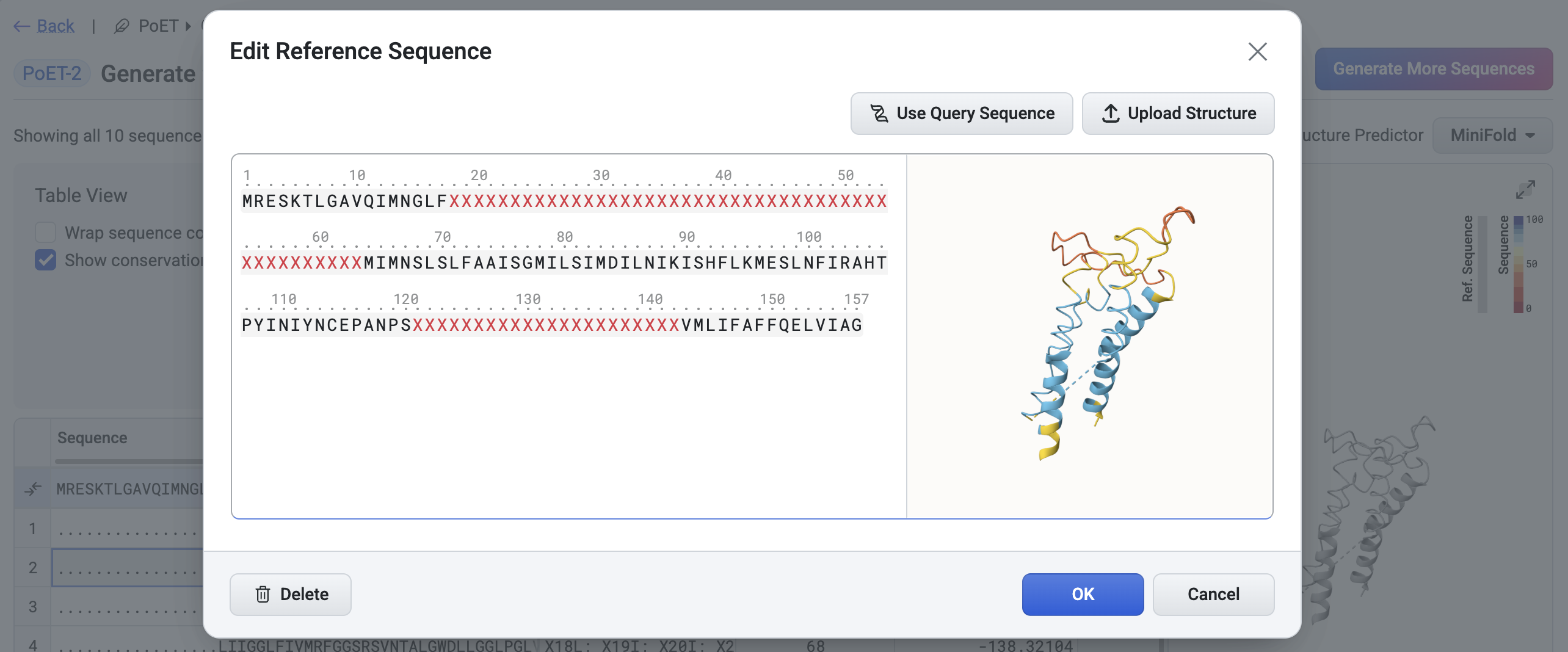
By default, the query structure is used as the reference structure for structural comparisons. You can change it by selecting an alternative structure via the Edit Reference Sequence button, accessible from the results table Settings in the results table.